
Conférence sur les thérapies de la maladie de Huntington 2018 – jour 3
Mises à jour du jour 3 de la Conférence sur les Thérapies de la Maladie de Huntington : la protéine huntingtine – et sa réduction

Bonjour depuis le dernier jour de la Conférence sur les Thérapies HD 2018 ! Deux sessions aujourd’hui, la première centrée sur la protéine produite par le gène HD. La seconde inclut des mises à jour sur les Essais de Réduction de la Huntingtine de Wave Life Sciences et Ionis Pharmaceuticals.
Jeudi matin – protéine huntingtine
Chaque patient HD a hérité de la même mutation – un allongement de la séquence C-A-G. Cette expansion se produit dans un gène que nous appelons maintenant le gène HD. Les gènes sont utilisés par les cellules comme instructions pour fabriquer des protéines – la première session d’aujourd’hui se concentre sur la protéine HD.

Sandrine Humbert, Université Grenoble Alpes, s’intéresse depuis longtemps au développement du cerveau et à la façon dont le gène et la protéine HD influencent ce processus. Pour comprendre ce processus, le laboratoire d’Humbert a créé une souris dont le cerveau était dépourvu du gène et de la protéine HD. Ils ont découvert que les cellules sans le gène HD se divisaient et se déplaçaient de manière anormale. Pendant le développement du cerveau, les cellules nouvellement nées rampent vers leur emplacement approprié, souvent en grimpant le long de ‘cordes’ formées par d’autres cellules. Ce processus est altéré lorsque le gène HD est supprimé, suggérant des rôles importants du gène HD dans ce processus.
Andrea Caricasole, IRBM Science Park, mène une étude à grande échelle sur les « modifications post-traductionnelles » de la protéine Huntingtine. Cela fait référence à de minuscules « décorations » chimiques de la protéine Huntingtine. Ces décorations permettent aux cellules d’ajuster la fonction des protéines. La protéine Huntingtine, par exemple, a probablement des dizaines de ces marqueurs qui sont ajoutés et retirés, ajustant la fonction de la Huntingtine en réponse à un large éventail de signaux. Beaucoup de ces décorations ont des effets intrigants sur la protéine Huntingtine, et peuvent même empêcher la protéine huntingtine mutante d’endommager les cellules. Nous en avons déjà parlé sur HDBuzz. L’équipe de Caricasole développe des tests très sensibles pour les décorations individuelles de la protéine Huntingtine. Cela leur permet de suivre lesquelles sont modifiées au cours de la maladie, et peut-être de chercher des moyens de les corriger.
Rohit Pappu, Université de Washington, adopte une approche très ciblée pour comprendre la protéine Huntingtine. Son laboratoire développe des outils pour étudier la partie de la protéine dont la forme est influencée par la mutation HD. Le laboratoire de Pappu utilise d’énormes ressources informatiques pour essayer de prédire la forme de la partie de la protéine huntingtine altérée par la mutation. Ces techniques leur permettent d’observer une forme de « têtard ». La forme de ce têtard a fait l’objet d’un débat intense dans le domaine de la HD ! Les techniques de Pappu soutiennent fortement un côté de ce débat, ce qui aidera sûrement à mieux comprendre cette partie cruciale de la protéine huntingtine
Xaio-Joang Li de l’Université Emory a développé un modèle de souris intéressant où le gène de la huntingtine peut être désactivé chez les souris adultes, dans le cerveau, le corps ou les deux. Ces souris n’ont pas de gènes huntingtine étendus – elles nous aident simplement à comprendre si la désactivation de la version ‘saine’ du gène a des conséquences. De façon rassurante, lorsque le gène est désactivé, rien de grave ne se produit dans le cerveau. De manière inattendue, la désactivation du gène a produit une inflammation du pancréas. On ne sait pas ce que cela pourrait signifier pour les patients, mais les traitements actuels de réduction de la huntingtine ne devraient pas réduire significativement les niveaux de huntingtine dans le corps – seulement dans le cerveau. Li a également utilisé l’édition génomique CRISPR-Cas9 pour supprimer la partie nocive du gène HD chez les souris. La désactivation du gène mutant chez les souris a réussi à réduire la formation de la protéine huntingtine toxique et les souris se déplaçaient mieux aussi. Dr Li a été très occupé ! Il a également créé un modèle porcin de la maladie de Huntington en utilisant l’édition génomique CRISPR. Cela pourrait être utile pour tester de nouveaux médicaments car le cerveau du porc est similaire à celui de l’humain.
« Kochanek a congelé la protéine et utilisé un faisceau d’électrons pour prendre des milliers de photos. Celles-ci ont ensuite été combinées par ordinateur pour produire les premières images de la structure moléculaire détaillée de la protéine huntingtine. »
Ankur Jain de @UCSanDiego étudie l’ARN – les « molécules messagères » générées lorsqu’une cellule veut utiliser les instructions de l’ADN pour fabriquer une protéine. Notre ADN vit dans le noyau de nos cellules, mais l’ARN flotte librement dans toute la cellule. La façon traditionnelle de penser à de nombreuses maladies cérébrales génétiques est qu’elles sont causées par des protéines toxiques, mais il y a de plus en plus de preuves que parfois les molécules d’ARN messagères produites à partir de gènes mutants peuvent aussi être toxiques. Par exemple, certaines séquences d’ARN peuvent se coller à d’importantes machines protéiques et les empêcher de faire leur travail dans la cellule. Un signe possible d’ARN toxique est la formation de taches anormales d’ARN observées dans les cellules dans la HD et d’autres maladies cérébrales. Jain a découvert qu’il peut former des taches artificielles d’ARN en le chauffant et en le refroidissant comme de la gelée. Ces taches ne se forment que lorsque l’ARN contient des séquences collantes comme celle de la séquence CAG dans la HD. On ne sait pas si ces taches d’ARN causent des dommages dans la HD, mais c’est possible. Par exemple, si l’ARN est coincé dans le noyau, il ne peut pas être utilisé pour générer des protéines. Les molécules antisens (similaires à celles actuellement utilisées dans les essais humains HD) peuvent se coller à l’ARN dans le noyau et les empêcher de former des taches. D’autres médicaments pourraient théoriquement être utilisés pour s’attaquer au problème de la viscosité de l’ARN dans les maladies cérébrales aussi.
Présentation de dernière minute passionnante maintenant de Stefan Kochanek, dont le laboratoire vient de découvrir la structure de la protéine huntingtine ! Découvrir à quoi ressemblent les protéines est une étape vraiment importante pour comprendre comment elles fonctionnent et comment modifier cela avec des médicaments. Le gène de la huntingtine a été découvert il y a 25 ans mais la protéine est grande, instable et collante, ce qui a rendu sa structure très difficile à découvrir. Une équipe a même envoyé la protéine dans l’espace pour essayer de former des cristaux, mais hélas, sans succès. L’équipe de Kochanek a réussi là où d’autres ont échoué et leurs résultats viennent d’être publiés dans Nature. La grande percée a été la stabilisation de la huntingtine en utilisant une autre protéine appelée HAP40 (« protéine associée à la huntingtine 40 »). Une fois stabilisée avec HAP40, Kochanek a congelé la protéine et utilisé un faisceau d’électrons pour prendre des milliers de photos. Celles-ci ont ensuite été combinées par ordinateur pour produire les premières images de la structure moléculaire détaillée de la protéine huntingtine. C’est vraiment cool et nous donne beaucoup de matière à travailler. Une mise en garde cependant : certaines zones étaient encore trop instables pour déterminer la structure – y compris la partie cruciale au début de la protéine qui contient la mutation.
Jeudi après-midi – réduction de la huntingtine
Grande fin de journée et de la conférence avec la session sur les thérapies de réduction de la Huntingtine qui commence. La réduction de la huntingtine fait référence aux approches qui visent à réduire les niveaux de la protéine Huntingtine. Il y a beaucoup de façons de faire cela, mais beaucoup d’entre elles ciblent l’ »ARN » qui est un intermédiaire entre l’information dans le gène HD et la protéine Huntingtine.
Michael Rape, UC Berkeley, s’intéresse à tromper les cellules pour qu’elles détruisent des protéines individuelles dans une cellule. Dans de nombreux cas, y compris la HD, il serait vraiment utile de supprimer sélectivement une protéine spécifique. Les cellules ont plus d’une voie de dégradation des protéines – une voie importante utilise une minuscule décoration chimique appelée « ubiquitine » comme étiquette. Les cellules reconnaissent l’ubiquitine comme une sorte de signal « mangez-moi » et décomposent les protéines qui les portent. Le laboratoire de Rape a été impliqué dans la compréhension de la façon dont les cellules utilisent les étiquettes d’ubiquitine pour marquer les protéines qui doivent être détruites très rapidement – une qui pourrait être toxique, par exemple. Le laboratoire de Rape a construit des outils qui permettent aux chercheurs, pour la première fois, d’observer les protéines passer par cette voie de destruction rapide. La machinerie de destruction rapide des protéines est un outil puissant – un que le laboratoire de Rape souhaite exploiter. Une technique récemment développée – appelée « PROTAC » – permet aux chercheurs d’exploiter le système de l’ubiquitine pour pousser les cellules à détruire des protéines spécifiques.
Scott Zeitlin (Université de Virginie) travaille avec des souris HD pour essayer de comprendre ce qui se passe lorsque nous réduisons la huntingtine mutante, la huntingtine normale ou les deux. Gardez à l’esprit que chaque personne hérite d’une huntingtine de chaque parent – et la plupart des personnes atteintes de HD ont une copie normale et une copie mutante. Les scientifiques appellent la protéine saine / normale « de type sauvage » car c’est celle qui est la plus commune dans la nature. Ces questions sont importantes car toutes les thérapies de réduction de la huntingtine visent à réduire la quantité globale de protéine huntingtine présente dans le cerveau. Certaines, comme le médicament d’Ionis, réduisent les deux versions de la protéine de manière égale. D’autres, comme les médicaments de Wave, cherchent à réduire la protéine mutante plus que la protéine de type sauvage. Nous pensons qu’il est probable que la réduction de la protéine mutante seule ou en parallèle avec la protéine de type sauvage sera bénéfique – mais c’est encore une question ouverte de savoir si la réduction de la huntingtine est sûre. Zeitlin a élevé des souris chez lesquelles la production de la protéine mutante, de type sauvage ou des deux peut être réduite après que la souris a complètement grandi. Zeitlin a constaté que la réduction précoce de la huntingtine mutante avait un plus grand effet en termes d’accumulation de la protéine dans le cerveau. De même, la réduction précoce de la huntingtine mutante avait de plus grands bénéfices sur la perte de poids et la capacité de mouvement chez les souris. Il en était de même pour la réduction de la production des deux versions de la protéine – le traitement précoce avait de plus grands bénéfices. En résumé : plus tôt c’est mieux quand il s’agit de réprimer la huntingtine. Sur un test (force de préhension), la réduction de la seule protéine mutante a amélioré les performances, mais la répression des deux versions ne l’a pas fait. Sinon, les deux approches étaient à peu près aussi efficaces et le facteur clé était la précocité du traitement. Zeitlin a également examiné ce qui se passe si on laisse la huntingtine rebondir, et c’était mauvais pour les souris. Cela suggère qu’un traitement à long terme est meilleur qu’un traitement à court terme – exactement ce à quoi on s’attendrait.
Jodi McBride, OHSU, décrit son travail utilisant des virus inoffensifs pour délivrer des instructions aux cellules cérébrales qui les aident à fabriquer leurs propres molécules détruisant l’ARN. L’un des avantages de ce type d’approche est que les virus permettent aux molécules détruisant l’ARN d’être produites indéfiniment, permettant en théorie un traitement unique. McBride étudie son traitement en le délivrant à des singes, qui ont des cerveaux grands et complexes beaucoup plus similaires aux nôtres. Spécifiquement, son équipe travaille sur la délivrance du virus à une partie du cerveau appelée le « putamen ». Le putamen est particulièrement intéressant, car c’est l’une des régions cérébrales les plus vulnérables dans la HD – subissant une importante atrophie chez les personnes qui héritent de la mutation HD. McBride décrit les améliorations de la chirurgie cérébrale nécessaire pour délivrer les virus, y compris l’utilisation de l’IRM pour imager le cerveau pendant les injections. Le traitement viral a conduit à des réductions de l’ARN du gène HD d’environ la moitié dans tout le putamen, une amélioration notable par rapport aux tentatives précédentes. Ensuite – Mike Panzara de Wave Life Sciences, qui planifient 2 essais utilisant des « Oligonucléotides Antisens » (ASO) pour la HD. Les ASO sont de courts morceaux d’ADN modifiés qui entrent dans les cellules et détruisent un ARN cible, réduisant les niveaux de la protéine cible
Panzara dit à la foule que Wave mène actuellement deux essais d’ASO chez des patients HD. Pourquoi deux ? L’approche de Wave repose sur le ciblage de minuscules variations génétiques – appelées SNP, ou « snips » – dans le gène HD. Ces minuscules variations ne causent pas la HD, elles font simplement partie de la variation génétique normale entre les personnes – la raison pour laquelle nous ne sommes pas tous des jumeaux identiques. De manière intéressante, ces variants ne se trouvent que sur l’une des 2 copies du gène HD que chaque personne possède. En ciblant ces variants, les ASO de Wave peuvent distinguer entre les copies mutante et non mutante du gène HD. Wave mène actuellement des études de sécurité précoces de 2 ASO dans des études appelées PRECISION-HD1 et PRECISION-HD2. Les ASO utilisés dans ces études ciblent différentes variations génétiques dans le gène HD. L’astuce avec cette approche est que les gens doivent non seulement avoir hérité de la mutation HD, mais aussi des variants accompagnants qui permettent de cibler uniquement la copie mutante du gène. Donc ces essais sont nécessairement concentrés sur les patients portant ces variations. Wave a développé des technologies vraiment cool pour détecter ces variants, et déterminer lesquels sont sur la copie mutante du gène HD, pas la copie normale. Wave a mené une étude préliminaire dans laquelle ils ont pu trouver des cibles pour leurs ASO chez 64% des volontaires
« Applaudissements spontanés lorsque Tabrizi remercie les courageux volontaires de la première étude, les qualifiant de ‘véritables héros de la recherche’. »
Ensuite, Anne Smith d’Ionis et Sarah Tabrizi d’UCL présentent les résultats d’un essai conçu pour tester les ASO ciblant les deux copies du gène HD. C’est l’aboutissement de nombreuses années de travail – Smith rappelle à l’audience que le programme Ionis a commencé en 2005 ! Ils ont commencé par des études sur les cellules et les animaux, qui ont fourni des preuves précoces que les traitements ASO réduisent la protéine Huntingtine et améliorent les symptômes similaires à la HD. En 2012 et 2013, les résultats des études sur le modèle de souris HD ont été publiés, démontrant que la réduction de la huntingtine améliorait les symptômes similaires à la HD. Smith expose la logique qu’@ionispharma a utilisée pour prendre la décision d’utiliser des ASO ciblant les deux copies du gène HD, plutôt que juste la copie mutante. Un avantage des ASO est qu’ils se distribuent largement dans tout le cerveau. Smith montre des données d’expériences sur les singes démontrant qu’après injection dans le liquide céphalo-rachidien, les ASO se distribuent très largement dans tout le cerveau. Ionis a également étudié la distribution chez des animaux encore plus grands, comme les porcs, trouvant que le médicament se distribuait très largement. Des études de toxicité ont ensuite été menées, suggérant que l’administration à long terme du médicament était très bien tolérée (jusqu’à 15 mois dans les études sur les singes). Il est presque impossible de prélever du tissu cérébral chez les patients traités avec des ASO – alors comment saurons-nous si l’ASO a fait son travail ? Smith décrit des études sur les singes établissant une relation entre la réduction de la huntingtine dans le cerveau et la réduction dans le liquide céphalo-rachidien. Cela a permis à Ionis de construire un programme informatique très compliqué pour prédire combien de réduction de la Huntingtine se produit dans le cerveau et le liquide céphalo-rachidien, qui est facilement accessible par une ponction lombaire. À ce stade, Ionis a été rejoint par un grand partenaire pharmaceutique, Roche qui a les ressources et l’expérience pour mener des essais humains compliqués pour les ASO.
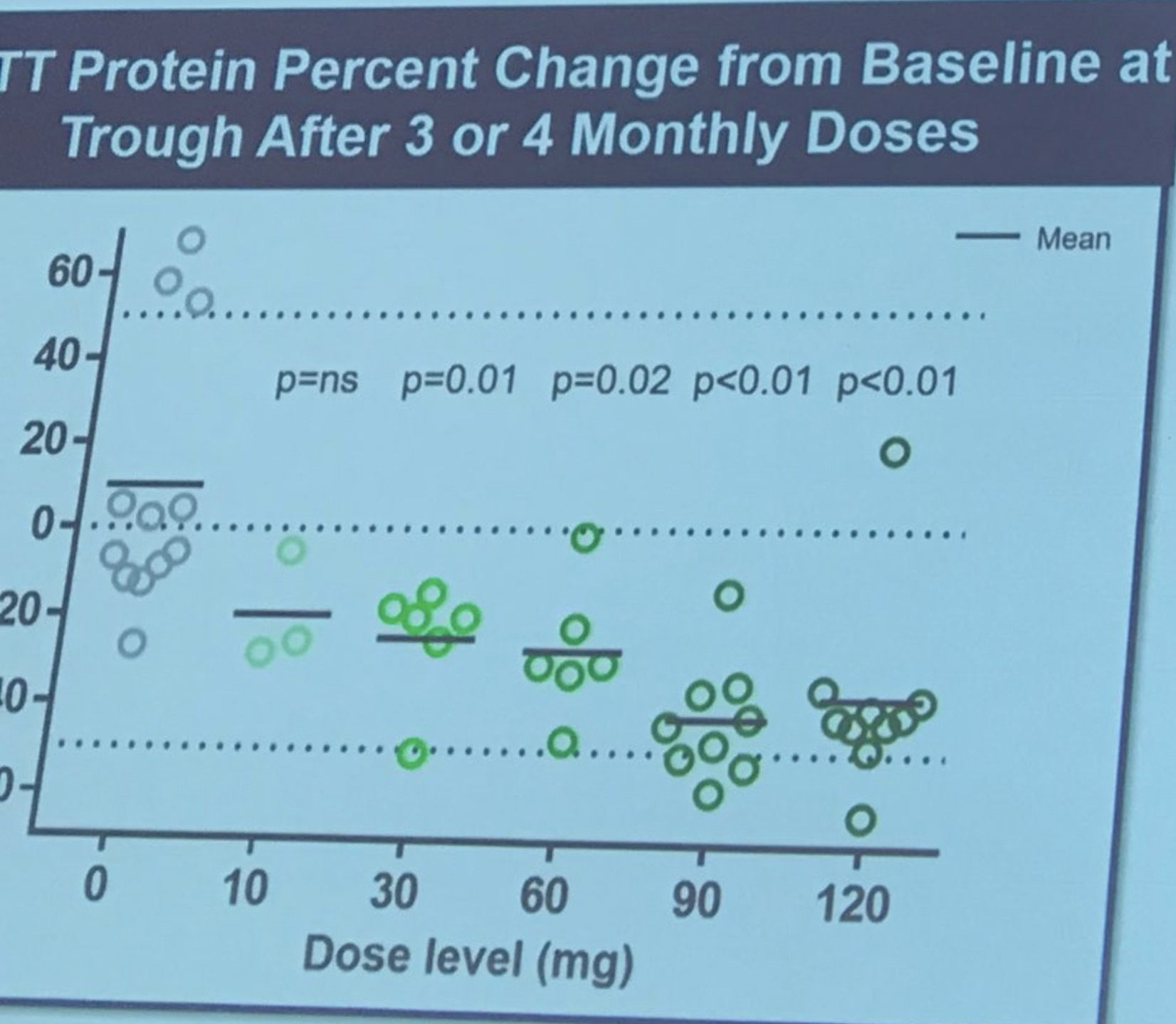
Sarah Tabrizi prend la parole pour décrire le premier essai humain du traitement ASO d’Ionis/Roche. Cette étude était une étude de « sécurité » – la raison principale de faire l’étude était d’établir si le médicament était sûr. L’étude a été menée dans 9 sites au Royaume-Uni, en Allemagne et au Canada. Les ASO ont été administrés aux patients par perfusion dans le liquide céphalo-rachidien avec une « dose croissante », ce qui signifie que les premiers sujets ont reçu une faible dose et les sujets suivants une dose plus élevée. Cette augmentation prudente de la dose est faite pour permettre des évaluations de sécurité par des médecins indépendants de l’étude. Cette étude incluait 46 volontaires incroyablement courageux, qui étaient prêts à prendre le risque d’être les premières personnes à être exposées au médicament. Les chercheurs ont pu mesurer les niveaux de la protéine Huntingtine dans le liquide céphalo-rachidien – qu’ils avaient précédemment montré comme étant très bien corrélés avec les niveaux cérébraux (que, rappelons-le, nous ne pouvons pas mesurer directement).
L’ampleur de la réduction est vraiment frappante – en moyenne, jusqu’à 40 à 50 % ! Tabrizi décrit le sentiment des chercheurs que la réduction de la huntingtine pourrait continuer à s’améliorer pendant près de 6 mois. Et voici à quoi, selon les prévisions de Tabrizi, cela correspond en termes de réduction des protéines cérébrales. Ionis a mis au point une sorte de modèle qui lui permet de faire des prédictions sur la relation entre la réduction de la huntingtine dans le liquide céphalo-rachidien et dans les tissus cérébraux. Cela suggère que la réduction de la huntingtine dans les tissus cérébraux pourrait être assez importante. Les patients ont été surveillés très attentivement pour assurer leur sécurité, et aucun événement indésirable majeur n’a été constaté. Tabrizi – « Le médicament était sûr et bien toléré à toutes les doses testées ». Succès ! Tous les sujets de l’étude participent maintenant à ce qu’on appelle une « extension ouverte » – ceux qui recevaient le placebo sont passés au médicament et continueront à être surveillés. Applaudissements spontanés lorsque Tabrizi remercie les courageux volontaires de la première étude, les qualifiant de « véritables héros de la recherche ».
Quelle belle façon de terminer la réunion ! Des moments incroyablement excitants nous attendent, car Roche et Ionis prévoient le prochain essai, qui sera conçu pour déterminer si le médicament améliore les symptômes de la maladie de Huntington chez un plus grand nombre de personnes.
Mise à jour : Déclaration d’Ionis à la communauté sur les résultats.
En savoir plus
Pour plus d’informations sur notre politique de divulgation, consulte notre FAQ…


